
Stephan Guyenet's Blog
May 11, 2021
Mistakes page for The Hungry Brain
I make mistakes, and acknowledging and correcting them is an important part of how I grow. On this page, I list every mistake I know of in my book The Hungry Brain, and whether they have been corrected in the paperback edition. Thanks to everyone who has pointed out mistakes!
I’m certain there are more I haven’t caught yet. If you spot one, please let me know and I’ll add it to the list.
P. 44. The book refers to the psychological concept of punishment as “negative reinforcement”. Negative reinforcement is a psychological concept as well, but not the one I intended to refer to in that passage. This has been corrected in the paperback version. Pointed out by Rebecca Strickland.P. 51. The book states that “Humans have a strong preference for salt (sodium chloride)– one inclination we don’t share with rats.” I discuss this further in the associated footnote. I don’t think this is a mistake exactly because rats don’t enjoy salty food like humans, but Anthony Sclafani pointed out to me that rats do prefer to drink a dilute salt solution to plain water. I removed the statement in the paperback.P. 54. In a footnote, the book states that the Diagnostic and Statistical Manual of Mental Disorders is published by the American Psychological Association. It’s actually published by the American Psychiatric Association. This has been corrected in the paperback version. Pointed out by Sylvia Karasu.P. 136-7. While discussing a rat study by Barry Levin and Ambrose Dunn-Meynell, I made two errors in describing the experimental diets. First, I stated that the researchers “restricted their food intake while keeping them on the palatable diet”. In fact, the rats were switched back to unrefined rodent chow during the restriction period. Second, I stated that “they quickly bounced back up to the weight of rats that had been eating an unrestricted palatable diet and gaining weight the entire time”. In fact, the latter group was placed on an unrestricted palatable diet for 12 weeks, then unrestricted unrefined rodent chow for an additional 16 weeks, and were on chow when the other group was switched and the two groups converged in weight. Neither error impacts the substance of the passage. Pointed out by a friend of Gary Taubes, via Gary. P. 171-2. I discuss a twin study of the genetic contribution to obesity conducted by Mats Borjeson and published in 1976, and the passage could be read as implying that he was the first to perform such a study. Twin studies on obesity were published prior to Borjeson’s study, as reviewed in the 1966 paper “A review of genetic and constitutional factors in human obesity“, by Carl Seltzer and Jean Mayer. I don’t recall who pointed this one out to me, but thanks, whoever you are.P. 175. I attribute the quote “genetics loads the gun, and environment pulls the trigger” to Francis Collins. Although Collins said it, the quote is typically attributed to obesity researcher George Bray. This has been updated in the paperback version. Pointed out by Sylvia Karasu. Tweet Share on Tumblr
Share on Tumblr



February 1, 2021
Does energy imbalance (calories in > out) cause obesity?
Gary Taubes argues that energy imbalance (calories in > out) does not cause obesity; rather, energy imbalance is a result of the fattening process. In this post, I evaluate this claim, which boils down to the question of which side of the energy balance equation is driving the other. A version of this debate has been happening within the obesity research community, including my corner of it, for decades. The truth is that while fat mass can clearly be changed by altering energy balance, and energy imbalance is required for obesity to occur, we don’t know for sure which side of the equation drives the development of common obesity. Current evidence suggests that energy imbalance probably drives obesity development, but the body fat regulatory system may drive energy imbalance and contribute to obesity development as well. Regardless of this, the evidence suggests that the primary body system in charge of the energy balance equation as a whole is probably the brain, not insulin or fat tissue.
I recently had a spirited debate on Twitter with journalist and low-carb diet advocate Gary Taubes about the energy balance equation. The debate relates to a long-standing difference of opinion about the implications and usefulness of the equation. Taubes’s arguments about energy balance have been persuasive to the general public, and a few researchers, so I think it’s worthwhile to address them. This was also an opportunity for me to clarify my own thinking.
What is the energy balance equation, and what is the claim about it?The energy balance equation states that changes in the amount of energy in the body are equal to the amount of energy entering the body, minus the amount leaving:
Change in body energy = Energy in – Energy out
This equation is an expression of the first law of thermodynamics, it’s simple arithmetic, and it’s not in question. Since fat tissue is by far the most capacious and most modifiable energy storage site in the body, substantial changes in the energy content of the body are represented by changes in body fat mass.
The body of a person with mild obesity contains about twice as much energy as the body of a lean person of the same sex and height, and this difference in body energy content is accounted for almost exclusively (~92%) by differences in fat mass.* It is of obvious interest to understand how this extra energy arrived in the body.
On many occasions, including in his new book The Case for Keto, Taubes has expressed a dim view of how he believes the energy balance equation is interpreted by researchers:
As sceptics pointed out at the time, though, the energy balance notion has an obvious flaw: it is tautological. If we get fatter (more massive), we have to take in more calories than we expendâ thatâs what the laws of thermodynamics dictateâand so we must be overeating during this fattening process. But this tells us nothing about cause. Hereâs the circular logic:
Why do we get fat? Because we overeat.
How do we know weâre overeating? Because weâre getting fatter.
And why are we getting fatter? Because weâre overeating.
And so it goes, round and round
Gary Taubes. BMJ 346:16-17. 2013.
The key claims in this passage are that the energy balance equation tells us nothing about what causes obesity (“this tells us nothing about cause”), and assuming that energy imbalance causes obesity because of the energy balance equation is illogical circular reasoning (“here’s the circular logic”).
The first thing that stands out to me is that I can’t recall having encountered in the scientific literature a circular argument like the one Taubes makes. I certainly don’t recognize it as a pervasive or fundamental way of thinking in my field. The common argument is that the energy balance equation implies that energy in > out causes obesity. That may be debatable but it isn’t circular. Taubes’s version is circular because he tacks on a series of self-referencing assertions that aren’t part of the original argument. In his telling, researchers answer questions about obesity by making assumptions that are based on one another.
Fortunately, researchers have a better way of answering questions than making circular assumptions: experimental evidence. For example, we don’t have to assume anything about energy intake– we can measure it. That’s how we know it’s typically higher in people with obesity. See this graph of accurately measured energy intake graphed against body mass index of 15 men of widely varying body fat levels.**

Likewise, we don’t have to assume anything about the impact of increasing energy intake on body fatness– we can measure it. That’s how we know increasing energy intake increases body fatness, regardless of whether the excess energy comes from carbohydrate or fat.
In the end, I don’t know what to make of Taubes’s circular argument. It’s obviously illogical but I don’t recognize it from the scientific literature, and it ignores quite a bit of relevant scientific evidence. I’m not sure who it’s arguing against. Maybe I’m missing something?
Nevertheless, I think there’s a logical idea in the passage that gets at an interesting question. So I’m going to steel-man it, which means to present the strongest, most charitable version of it that I can. Here goes:
People often focus on the right side of the energy balance equation (energy in/out) as driving the left side (change in body energy). “She ate more, therefore she gained fat”.However, the equation itself doesn’t imply directionality.It’s possible that this is causally flipped in the development of common obesity, where the original cause is a biological process that drives fat gain, and energy imbalance is simply a tool serving that process. “Her body was ‘trying’ to gain fat, and this process caused her to eat more and/or reduced her metabolic rate”.If the latter scenario is true, it would be more appropriate to apply the word “cause” to the biological process rather than energy imbalance, since the former is ultimately driving the latter.An analogy Taubes uses frequently is that of growth. In the case of a child becoming an adult, wouldn’t we say that the growth process is causing the child to eat more, rather than eating more causing the child to grow?
The argument is logical, but that doesn’t necessarily mean it’s correct in the context of common obesity. Let’s evaluate it.
Energy imbalance is causal, but that doesn’t necessarily mean it’s in chargeAs mentioned previously, the body of a person with mild obesity contains about twice as much energy as the body of a lean person, and nearly all (~92%) of the excess energy is in fat tissue.*
According to physics and simple arithmetic, as expressed by the energy balance equation, this energy accumulation must result from energy imbalance (energy in > out). There is no other possible way for the extra energy to arrive in the body. Energy imbalance is required for the state of common obesity to develop. It is therefore causal, as opposed to just being an innocent bystander. That said, I acknowledge that there is an important semantic issue around how we use the word “cause”. I’ll come back to that.
Energy imbalance is also causal in the case of a child growing, because an adult’s body contains more energy than a child’s body. The normal transition from child to adult cannot occur without energy imbalance. But growth isn’t a very good analogy for obesity. One reason is that in obesity, excess energy in the body is the condition. The difference in the energy content of a lean body vs. an obese body is nearly all accounted for by the difference in fat tissue mass, which is the definition of obesity. That isn’t the case in development. A second reason the analogy fails is that increasing a well-nourished person’s calorie intake causes them to gain body fat, while increasing a well-nourished child’s calorie intake does not cause them to develop into an adult. Similarly, reducing calorie intake causes body fat loss but doesn’t cause an adult to regress back to a child. I won’t consider this analogy further.
I think the best way to think about obesity, and everything else, is as a causal chain (or causal web if you want to be fancy). Everything is caused by something, so whatever our variable of interest (e.g., body fatness), it was caused by one or more upstream factors, which in turn were caused by something else, ad infinitum. Each cause is a link in the chain. No matter what your model of obesity is, in order to explain the development of an obese body, energy imbalance has to be a link in the model’s causal chain. If it isn’t, you’ve run afoul of physics and arithmetic.
That said, I do think Taubes’s argument has some logic, and it comes down to the question of what is ultimately controlling the system. In a typical causal chain, there are some links that actively determine the system’s output, and other links that just transmit the signal. Another way of thinking about this is that some links “give orders” and other links just “take orders”. The logical piece of Taubes’s argument is that the energy balance equation itself doesn’t imply which side is giving orders, and which is taking orders.
Consider this example. The speed of a car is determined (in large part) by the amount of force its tires exert on the road. Tires cause a car to move forward. Yet if we want to understand why some cars tend to drive faster than others on a local highway, studying their tires won’t provide any insight into that: the tires are just taking orders from upstream links in the causal chain (axle, transmission, engine, accelerator, etc.). To understand why some cars drive faster than others, we have to focus on the part of the system that’s giving orders, which is the person behind the wheel.
But what do we apply the word “cause” to? This might seem like splitting semantic hairs, but I think it’s actually at the heart of this debate. Is every link in the causal chain a “cause”? Or does a link have to give orders to be a “cause”? Taubes appears to use the latter definition, others often use the former definition, and this causes the two sides to talk past one another. I believe this semantic miscommunication explains a substantial part of the disagreement.
I’ll give an example to make this more concrete. Here is a passage from a post on Taubes’s website, in which he quotes from a review paper written by my scientific mentor Mike Schwartz:
âObesity, by definition, results from ingesting calories in excess of ongoing requirements,â Schwartz and his co-author write, and Iâm arguing that any researcher who makes this statement for any reason other than to make the point that itâs meaningless and misleading is living and working in an energy balance paradigm â calories-in, calories-out.
Taubes interprets this passage as implying that energy imbalance is giving orders, leading him to heap contempt on it. Yet if one reads the paper, it’s about the biological regulation of fat mass and energy balance. Furthermore, the paper explicitly discusses the question of whether energy imbalance leads or follows in the development of common obesity, and concludes that the question is unresolved***.
Clearly, Taubes’s interpretation of the passage is not what the authors intended. The reason for the miscommunication is that the paper is making the uncontroversial observation that energy imbalance is a link in the causal chain of obesity, while Taubes interprets it as a statement about which side of the equation is giving orders. Taubes does this a lot, but in fairness I think the research community is often guilty of thinking/communicating less than optimally on this issue. In this particular case the context makes the intention clear, but that isn’t always true.
Extending the car analogy to obesity, the steel-man version of Taubes’s argument would state that yes, energy imbalance is in the causal chain leading to obesity, but it’s just taking orders. The left side of the energy balance equation– a biological process driving fat mass expansion– is what’s giving orders. That’s the theory, but is it correct?
What’s giving orders, and what’s taking them?The first thing I’d like to point out is that the left and right sides of the energy balance equation could both be giving orders, and both be taking orders. The two possibilities aren’t mutually exclusive. And I think you can make a case for it going both ways. But even if the left side of the equation is giving orders, that doesn’t necessarily mean Taubes’s insulin-adipose mechanism is the explanation for it.
The second point I’ll make is that it can be difficult to discern which links in the causal chain are giving orders and which links are simply taking orders. In my opinion, this is a major challenge in scientific research. Let’s get back to the car analogy. Imagine that I conduct a study in which I pop the tires of one group of cars, but not a second group of cars, then I measure how fast the two groups drive. Examining the results, I might conclude that tires do in fact control the speed of a car, and therefore that tires are what determine why some cars tend to drive faster than others on my local highway. But even though my experiment demonstrated that tires are in the causal chain, my conclusion would be wrong because I haven’t demonstrated that tires give orders in the context I’m trying to understand.
Similarly, the examples that Taubes and others use to argue that insulin and adipose tissue “give orders” from the left side of the energy balance equation, rather than “taking orders” from the right side, are generally not from the condition of interest, common obesity. They’re from situations in which insulin signaling or adipose tissue function are pushed outside their normal operating parameters by experimental manipulation or disease, much like a car with popped tires: type 1 diabetes, insulin injection, mice with missing insulin genes or genetically manipulated adipose tissue function, etc. These experiments demonstrate that insulin and adipose tissue are in the causal chain of obesity, but not necessarily that they give orders in the context of regular people gaining weight.
When we examine the natural context– people living their normal lives and getting fatter or not– the picture suggests that insulin and adipose tissue are predominantly taking orders rather than giving orders:
In people living their normal lives, circulating insulin level, and the insulin response to carbohydrate, don’t reliably predict who will gain weight and who won’t. A person with high insulin tends to gain about the same amount of weight over the years as a person with low insulin. It’s hard to understand how elevated insulin could be the primary cause of obesity, given these findings. That said, if insulin were playing a minority role it could be hard to detect in this type of study.The most unbiased and informative way I know of determining what’s “giving orders” in a natural context is human genetics research. Genome-wide association studies (GWAS) tell us which locations in the genome are correlated with specific traits in people living their normal lives, and examining the functions of the genes that happen to pop up tells us which systems are “giving orders”. For example, the genes that correlate with type 2 diabetes risk are primarily related to insulin signaling and the pancreas. This tells us that to a large degree, insulin signaling and the pancreas are the biological systems in charge of type 2 diabetes risk. The genes that correlate with height are related to growth of the skeletal system and connective tissues. In obesity, the genes that correlate with body fatness are predominantly related to the brain, not to insulin or adipose tissue. This suggests that in the case of common human obesity, the brain is the primary body system that’s giving orders, and insulin and adipose tissue are primarily taking orders.This shouldn’t be particularly surprising, since the brain is already known to control both sides of the energy balance equation: it generates eating behavior, including what and how much we eat. It generates physical activity. It regulates metabolic rate. And it’s the only organ in the body known to contain a regulatory system for fat mass.
The brain is the body’s professional information-processing organ and it performs many functions that evolved to satisfy our ancestors’ needs related to energy and food. We can divide these into “homeostatic” and “non-homeostatic” functions that map onto the left and right side of the energy balance equation, respectively. Any control system can fail when placed outside its normal operating parameters, and these systems didn’t evolve for the modern context. Both homeostatic and non-homeostatic brain functions tend to favor overconsumption and maladaptive body fat gain in today’s world, as I describe in my book.
However– importantly– this still doesn’t tell us which side of the energy balance equation is giving orders. It remains possible that the left side is giving orders, but the brain is in charge of it. In fact, this is what people in my corner of the obesity research world (energy homeostasis) have been arguing might be happening for decades: the brain is in control of energy balance, so we should try to understand how that regulatory process is altered in obesity.
How do we answer this question? Let’s consider it from another angle. If energy balance is giving orders rather than taking them, we should be able to bring people from the obese state to the lean state, and from the lean state to the obese state, by changing energy balance alone.
Many studies (and many peoples’ experiences) have shown that reducing calorie intake causes fat loss, and if the cumulative calorie deficit is large enough, people can lose quite a bit of fat. At first glance, this seems like a home run for the argument that energy imbalance gives the orders. Certainly, there’s no doubt that energy imbalance has a powerful impact on fat mass!
However, when we look closer, the situation isn’t quite so cut and dried. The truth is that a person who has lost a substantial amount of fat is not the same as a person who had that leaner body all along. A weight-reduced person has a greater tendency to gain weight/fat, and when we look at this from the perspective of energy balance, we see that they have a hard time maintaining the lower calorie intake, and their metabolic rate has also slowed a bit more than their change in body mass would predict. In extreme cases, such as contestants on the weight loss game show The Biggest Loser, the decline in metabolic rate can actually be quite large. (As an aside, the tendency of a weight-reduced person to regain fat occurs in a state of reduced circulating insulin level.)
This is a phenomenon I call the “starvation response”, and it happens because body fat mass is biologically regulated. Current evidence suggests this regulation happens via the fat-regulating hormone leptin acting predominantly in the brain. Consistent with the genetics data, this is the only known system in the body that regulates fat mass. In my book The Hungry Brain, I review the research on this, which stretches back 180 years but has intensified since the discovery of leptin in 1994.
The “starvation response” to weight loss is an example of the left side of the energy balance equation giving orders, in addition to taking orders. When fat mass declines, there is a reaction from the body that seeks to modify energy balance in order to regain fat.
The situation is similar for fat gain. Increasing energy intake causes fat gain in scientific studies, but it’s hard to maintain substantial gains because the appetite reacts, pushing back against the change in energy balance. Whether or not fat gain via experimental overfeeding also causes a disproportionate increase in metabolic rate is controversial. It’s not difficult to cause a person to gain fat via overfeeding, but it seems to get more difficult the more you want them to gain, presumably because the body fat regulatory system “pushes back”. To some degree, this system protects against both fat loss and fat gain, but it’s obviously less effective at protecting against long-term fat gain in most people.
Insulin does not appear to play a key role in this process because overfeeding carbohydrate vs. fat has about the same effect on fat gain, despite different effects on insulin. Unlike the “starvation response”, the mechanism behind resistance to weight gain remains unclear, and is an active subject of research.
From all this, it’s clear that both sides of the energy balance equation can give orders that affect fat mass, and I think it makes a strong case that both sides do give orders in the context of voluntary weight loss. Yet, is experimental overfeeding an accurate model of the typical fattening process, or are we just popping tires? Does that scenario actually represent the process that causes common obesity in the real world? Here’s the truth: we don’t know for sure.
What we do know is that obesity consists of both a change in energy balance and a change in the biological system that regulates body fatness. Does energy imbalance cause fat gain, and the regulatory system adapts to higher fat mass over time, making it hard to slim back down? Or does the regulatory system go awry in the modern environment, driving fat gain and energy imbalance? Or both? Itâs a chicken-and-egg question that we donât yet have a definitive answer to.
We aren’t flying completely blind though. One way to examine this question is to see whether experimental overfeeding results in weight/fat gain that persists after the overfeeding period, or whether people shed all the excess weight. If people retain excess weight, it would imply that the regulatory system adapts to “defend” the higher level of fat mass, and in turn it would provide evidence that energy imbalance may be giving orders in the development of common obesity.
It turns out that people often do retain part of the excess weight/fat they gain during overfeeding. For example, Johannsen and colleagues overfed 35 young adults for 8 weeks, resulting in 17 pounds (7.5 kg) of weight gain and 9 pounds (4.2 kg) of fat gain. The researchers sent the volunteers home to live their normal lives, then measured their body weight and body composition six months later. At that point, they retained 43% of the excess weight they had gained during overfeeding, and a similar proportion of the excess fat.
This suggests that overfeeding had caused their body fat regulatory system to durably accept a higher “setpoint”. In turn, this implies that energy imbalance was probably giving the orders in this scenario. Some, but not all, other overfeeding studies have reported similar findings.
Experimental overfeeding might still seem like an artificial scenario, but consider that if we’re being honest, most of us “overfeed” ourselves periodically to some degree, meaning we eat/drink more than we need to because the food/drink is seductive, readily available, low-satiety, or for a variety of other reasons. Most annual weight gain in the US occurs during the 6-week holiday period, which represents only 12 percent of the year. On average, we lose some of that excess weight as we come out of the holiday period, but we retain some of it, to be built upon the next year. Our weight ratchets up a bit each year as a result of holiday eating, and the dynamics of it resembles a miniature version of experimental overfeeding.
This evidence isn’t definitive but it does suggest that energy imbalance probably gives orders in the development of common obesity, and therefore energy imbalance probably causes obesity under both definitions of the word “cause”. But this doesn’t exclude the possibility that the left side of the equation is also giving orders.
My guess is that both sides of the equation give orders in the real-life development of common obesity, but that the relative proportions depend on the person. But I don’t know for sure, and I doubt anyone does. This remains an important question for the research community. However, whichever side of the energy balance equation is giving orders, the brain appears to be the commander-in-chief of the equation as a whole.
I conclude the following:
Energy in/out is causal in obesity, but in and of itself, that fact doesn’t necessarily mean it’s giving orders. In terms of modifying fat mass, both sides of the energy balance equation are able to give orders, and also take orders. In the context of voluntary weight loss, both sides give orders.In the context of people developing common obesity in the real world, we don’t have definitive answers about which side of the equation is giving orders. The evidence suggests that energy imbalance probably gives orders, but we don’t know whether the other side of the equation does as well. I suspect it’s probably both sides and the proportion depends on the person, but that’s just a guess.Regardless of which side of the energy balance equation is giving orders, the brain appears to be the commander-in-chief of the equation as a whole.Insulin and adipose tissue probably take orders, for the most part, although I don’t think current evidence rules out a minority role for them in giving orders.Bonus: is the energy balance equation useful?To those who do obesity research, the value of the equation is obvious: it helps narrow down the possible field of explanations for the body composition of a person or lab animal, or changes in body composition. For example:
In people with obesity, energy intake and expenditure are typically higher than in people without obesity of the same sex and height. This tells us that common obesity is not the result of a reduced metabolic rate– a common but erroneous belief. It tells us that we should instead focus on mechanisms related to higher energy intake.The Hadza are a hunter-gatherer population in Tanzania, and they are characteristically lean. Although they have a higher physical activity level than a typical American, their overall energy expenditure is about the same as a typical American, after adjusting for the fact that their bodies are smaller and leaner. This tells us that higher energy expenditure due to exercise is not the reason they’re lean– we should instead focus on mechanisms related to lower energy intake.These are just two examples off the top of my head; the energy balance equation has many applications in obesity research. If you want to understand body fatness, it’s valuable to understand the energy balance characteristics of your subjects of interest.
AcknowledgmentsMany thanks to Kevin Hall and Karl Kaiyala for their helpful comments on a draft of this post.
Footnotes*This can be estimated using a few simple assumptions. First, the energy density of fat is 86.9 MJ/lb, and the energy density of lean tissue is 16.72 MJ/lb. Then, assume two men of average height (5’9″), one who is lean (BMI 22, 149 lbs, 12% body fat) and one who has mild obesity
(BMI 32, 217 lbs, 30% body fat). This yields total body energy contents of 3,746 MJ and 8,197 MJ. The body of the man with obesity contains 2.2 times more energy than the body of the lean man, and 92% of the excess energy is contained in fat tissue. The number is not 100% because people with obesity tend to have more lean mass than people who are lean.
**The correlation is strong at R2 = 0.77. Calorie intake was measured over 18 days in a metabolic ward in people who were weight-stable. Data are from table 1 of this study. A similar relationship between BMI and energy intake has been reported in many other studies that measured energy intake accurately but indirectly by measuring energy expenditure in weight-stable people. The first law of thermodynamics (conservation of energy) allows us to convert between energy intake and energy expenditure in weight-stable people.
***This passage from the paper illustrates that the authors do not assume energy imbalance is “giving orders” in the context of common obesity. They describe the question as unresolved:
Tweet
Another obstacle arises from a lack of scientific consensus regarding fundamental aspects of obesity pathogenesis. During caloric restriction, there is little question that reduced neuronal input from adiposity-related hormones activates responses (increased food intake, decreased metabolic rate) that favour recovery of lost weight. Whether the reverse is also trueâthat increased neuronal input from these hormones protects against weight gainâis hotly debated. Until this issue is resolved, the importance of several key observations, including CNS resistance to leptin and insulin documented in common forms of obesity, will remain uncertain. This is because if adiposity negative-feedback signals do not normally protect against weight gain, neuronal resistance to insulin and leptin cannot cause obesity. According to this view, the growing obesity epidemic can be attributed to an inherent lack of protection against obesity-promoting environmental factors, rather than to an underlying homeostatic disorder. Alternatively, if adiposity negative-feedback signals do, in fact, confer protection against obesity in normal-weight individuals, neuronal resistance to these signals must, by definition, favour weight gain, and unravelling the underlying causes takes on both pathophysiological and therapeutic urgency. As our understanding of normal and abnormal regulation of food intake and body adiposity grows in its sophistication, overcoming these obstacles will create new opportunities for therapeutic intervention.
Morton et al. Nature 443:289. 2006
Share on Tumblr
April 1, 2019
In a stunning reversal, archaeologists report that Paleolithic humans ate nothing but animal fat
A new study released today in the journal Nature upends our beliefs about Paleolithic peoples, suggesting that their diets may have been based solely on animal fat.
Previous research examining food particles in tooth plaque suggested that Paleolithic Homo sapiens and Homo neanderthalensis consumed a wide variety of plant and animal foods, leading to the belief that humans are natural omnivores. Yet new research suggests that while our ancestors may have chewed these foods, they probably didn’t swallow them.
Using the ratio of bismuth-84 to molybdenum-43 in six partially fossilized knee caps recovered from three European caves, a research team led by Nathan Hayes of the University of Pennsylvania was able to determine that the prehistoric humans ate no carbohydrate or protein at all, implying that their diet consisted entirely of fat. Based on bones and tools found in the same strata, the researchers believe these populations harvested the fat of large mammals such as aurochs, mammoths, elk, and wooly rhinoceroses, leaving the meat to rot.
“We are very confident in our conclusions”, said Hayes. “These people ate nothing but animal fat.”
The research community is stunned. Liu Zhang, a Yale biochemist, remarked “The implications of this finding go far beyond nutrition. It seems humans have a greater capacity to synthesize amino acids from fat than we thought.”
Hayes and his team believe the hunting grounds of Paleolithic humans in Europe were rich enough that they had no need to bother with inferior foods like plants and muscle tissue. Herds of mammoths were so thick during this time that it was almost impossible to throw a spear without hitting one in the carotid artery. Grains, legumes, fruit, and other plant foods were chewed for pleasure and to clean the teeth, but spat out rather than swallowed due to their inferior nutritional value.
Examining the kneecaps further, Hayes noted that the attachment points of their tendons were extremely robust. “These guys were ripped”, exclaimed Hays. “They definitely didn’t skip leg day. Or maybe it was all the fat they ate.”
April Fools!!
Tweet
Share on Tumblr
August 30, 2018
Quickly assessing the credibility of public experts
We live in a complex world, and it’s impossible to be an expert on everything that impacts our lives. In many domains, we have to trust the expertise of others to guide our decisions. Yet not all experts hold rational beliefs, and many people who are framed as experts in media are not actually experts. How do we separate the wheat from the chaff, focusing on high-quality sources of information and ignoring low-quality sources?
Over the years I’ve observed the behavior of various public figures in my areas of expertise, and I feel that I’ve developed pretty good nose for quickly sniffing out the credibility of public experts. In this post, I’ve attempted to take my intuitions and put them in writing so I can pass at least some of them on to others.
Here is a list of some of the heuristics I use when I’m assessing the credibility of people who are framed as experts. None of these are ironclad rules, but together they can help you get a quick sense of whether to listen to someone:
Does he have training or experience in the subject? This is particularly relevant when someone disagrees with expert consensus. Sometimes people get a platform just because they have a novel or interesting idea, even if that idea is unconvincing to a knowledgeable person. It is of course possible that the non-expert is right and the experts are wrong, but it’s unlikely. This is just a heuristic, since in some areas the experts are truly not knowledgeable. For example, economists are barely better than chance at predicting the short-term behavior of the stock market, but this doesn’t prevent some of them from prognosticating about it.
Does she try to explain everything with one idea? The work of Philip Tetlock, PhD and others has shown that people who have one big idea to explain everything (“hedgehogs” or ideologues) are very bad at accurately modeling the world, predicting outcomes, and recommending effective actions. These people are often selected for media attention because they are clear and confident about their beliefs. The world is a complex place, and people who are able to model that complexity in their minds have better information than those who aren’t. Look for people who tend to use multi-factor explanations.
Does he show nuance? Related to #2. There are usually exceptions and nuances. Is this person able to build them into his mental model?
Is she able to change her mind? Inability to change one’s mind is a sign of an irrational belief system. Can you find examples of this person changing her mind in the past when presented with new evidence or a better interpretation of existing evidence?
Does he lean toward conspiracy explanations and/or blaming the government? Large-scale conspiracies are improbable and the government is a favorite punching bag for cranks.
Does she cite evidence that isn’t representative of the literature as a whole? This is common among experts, less common among the best experts, and very common among non-experts. It’s also often easy to spot with a little effort. Do a quick Google Scholar search for a recent meta-analysis on the topic, ideally from the Cochrane Collaboration. Are the conclusions of the meta-analysis consistent with the evidence she is citing, or is she cherry-picking individual studies that support her position?
Does the topic relate to personal identity, or is it otherwise highly political or controversial? Domains such as religion, politics, and nutrition relate strongly to peoples’ personal identities. In this context, beliefs are often driven by group affiliation rather than rational consideration of evidence. The likelihood that someone is providing high-quality information in these domains is lower than in less controversial areas like physics or neuroscience.
What did I miss?
Tweet
Share on Tumblr
August 1, 2018
A more complete picture of the “collapse” of NuSI, and corrections to my previous post on it
After publishing my post Nutrition Science Initiative (NuSI) in retrospect, I received an email from Peter Attia, MD, the former president of NuSI, suggesting that I had gotten aspects of the story wrong due to a lack of additional context. Attia and I spoke twice by phone, and I think he has a point. I also had an additional conversation with Kevin Hall, PhD, one of the key researchers NuSI funded, for additional context from his perspective.
Salaries
NuSI executives didn’t set their own compensation, as I and others have implied. Compensation was set by the Compensation Committee of NuSI’s board, which did not include NuSI executives or employees. All overhead (i.e., all non-research dollars), including compensation, was paid directly from the Arnold Foundation grant with their knowledge and consent. According to Attia, donors other than the Arnold Foundation didn’t pay for salary or any other overhead; their funds went exclusively to research. Although Attia’s salary may seem high for a 501(c)(3) nonprofit, at its highest it was set following the recommendation of an external firm specializing in nonprofit executive compensation, at the request of the Board of Directors and with the approval of the Arnold Foundation. It seems that there was no self-dealing and my original post was incorrect in saying that NuSI executives “paid themselves enormous salaries.”
The collapse of NuSI
The reasons for the collapse of NuSI are more complex than what I portrayed in my article, and what appeared in the Wired article. A key reason NuSI collapsed is that it lost its funding, in part due to the fact that Attia was its most effective fundraiser and he left the organization at the end of 2015. Although Hall left NuSI’s Energy Balance Consortium (EBC) in 2016 due to scientific interference and personality conflicts, effectively shutting down the EBC at least temporarily, NuSI was larger than the EBC and the organization may have continued without it had funding not collapsed in 2016. The reasons why Attia left NuSI are complex, but they include undesired changes in his job requirements and personality conflicts.
Interference in the research process
NuSI did have its problems, some of which were described in my post and the Wired article, and some of which weren’t. I still believe NuSI tried to play a greater role in the research process than it should have, interfering with science and alienating certain researchers. Attia explained that from NuSI’s perspective, their intention was to create an adversarial collaboration that would be more effective at finding the truth than either camp alone. I actually think this is an admirable goal in many settings, and Daniel Kahneman, PhD describes a nice example of it in his book Thinking, Fast and Slow.
However, there are two reasons why I think it wasn’t a good idea in this particular case. First, this wasn’t the original agreement NuSI reached with EBC investigators. As Hall told me at the beginning of this saga in 2012, and reiterated in a recent conversation, the original agreement was that NuSI would fund studies on topics of interest and let the researchers take it from there. In other words, they would not be involved in the conduct, analysis, or reporting of the studies. Without this assurance, Hall wouldn’t have agreed to work with NuSI in the first place. However, after the EBC reported the results of its first study, which were unfavorable to the carbohydrate-insulin hypothesis, NuSI reneged on this agreement and began inserting itself into the research process in an increasingly assertive manner. The concept of adversarial collaboration emerged after these unfavorable findings and corresponded with NuSI’s desire to have greater control.
The second reason I think adversarial collaboration wasn’t a good idea in this case is that it was too dangerous due to the personalities, incentives, and strong beliefs of certain people on the NuSI side. This was a liability I flagged in 2012, which was apparent in the promotional materials NuSI published, and I think my concerns about this were realized. A key reason why I decided to support NuSI initially, despite this reservation, is that I was assured (as were Hall and his collaborators) it wouldn’t have control over the research process.
As I discussed in my initial article, NuSI did ask Energy Balance Consortium researchers to withdraw their abstract from the American Diabetes Association meeting, but this was because the researchers didn’t provide it to NuSI two weeks in advance, which their contract stipulated. While Attia argues that such agreements are common for this type of funded research, it still seems a little odd to me that NuSI actually enforced this rule and prevented the data (which challenge the carbohydrate-insulin hypothesis) from appearing at a major scientific meeting. Hall told me he wasn’t aware of this clause when he submitted the abstract. This incident would be less concerning if it weren’t part of a larger pattern of interference.
Conclusions
Although it still seems clear that NuSI interfered with the research process and alienated EBC researchers, I now believe the story of NuSI’s “collapse” is more complex than it initially appeared. I apologize to anyone who was harmed by inaccuracies in my initial article, particularly the salary issue. I’ll be annotating my original post to indicate the parts that I now think are wrong or misleading.
Tweet
Share on Tumblr
July 3, 2018
Why the carbohydrate-insulin model of obesity is probably wrong: A supplementary reply to Ebbeling and Ludwig’s JAMA article
I recently had the opportunity to collaborate with Kevin Hall, PhD and Rudy Leibel, MD on a commentary in JAMA Internal Medicine (1). It was fun for me to work with two researchers who I respect tremendously. Hall’s energy balance modeling work has brought important new insights to the obesity research field and Leibel is, well, the co-discoverer of leptin. And he has done as much as anyone else to help us understand how this hormone works in humans.
Our commentary is titled “The Carbohydrate-Insulin Model of Obesity Is Difficult to Reconcile With Current Evidence”, and we wrote it in response to a review paper in the same issue written by David Ludwig, MD, PhD and Cara Ebbeling, PhD, titled “The Carbohydrate-Insulin Model of Obesity: Beyond ‘Calories In, Calories Out'” (2). In this paper, Ludwig and Ebbeling lay out their argument for the carbohydrate-insulin model (CIM), as they refer to it. This is nearly identical to the model Gary Taubes advocates: obesity is primarily caused by the ability of carbohydrate to increase insulin secretion, which reduces levels of circulating fuels (glucose and free fatty acids), shunts fat into fat cells, and makes us fat, hungry, and sluggish. According to this model, high calorie intake and low calorie expenditure are a result of expanding fat tissue, not its cause. Ludwig and Ebbeling focus particularly on glycemic load, which is the degree to which diet impacts blood glucose. In my view, this review paper is the strongest defense of the model currently available.
While Ludwig and Ebbeling had ample space to develop their arguments, we were limited to 1,200 words and 8 references. So I’m going to add to our commentary here, expanding on some points and adding others. This post reflects my views and not necessarily those of Hall and Leibel, who were not involved in writing it.
I don’t want to be on the wrong side of history, and one way to do that is to make overly confident and categorical predictions. I still think it’s plausible that some insulin-related variable could be involved in obesity and/or fat loss, particularly 1) insulin resistance in energy-regulating circuits in the brain, and/or 2) blood glucose levels between meals, or some other signal of glucose availability. What I think is very unlikely to be correct is the hypothesis articulated by Ludwig, Ebbeling, and Taubes: the primary cause of obesity is carbohydrate-stimulated insulin acting on fat cells.
That said, I want to be clear that I think certain forms of carbohydrate are part of the explanation for obesity, and low-carbohydrate diets do cause fat loss in most people, with greater carbohydrate restriction typically resulting in greater fat loss. Like most diets, low-carbohydrate diets aren’t very effective against obesity in the average person, but they do have some effectiveness and they are certainly a valid tool in the toolbox. They may also be particularly useful for managing diabetes, although long-term outcomes remain uncertain.
Mischaracterization of opposing viewpoints
Ebbeling and Ludwig begin their JAMA piece by citing a recent Endocrine Society scientific statement written by leading obesity researchers, including Rudy Leibel and my mentor Mike Schwartz, MD (3). Ebbeling and Ludwig present it as an example of something they call the “Conventional Model”, which holds that calorie intake, expenditure, and body fatness are not biologically regulated and all we have to do is fire up our willpower and “eat less, move more” to conquer obesity. This is the opposite of what the Endocrine Society paper actually says, and our first clue is the fact that it’s published on behalf of the Endocrine Society in the journal Endocrine Reviews– both organizations dedicated to the study of hormones.
Schwartz, and most of the other authors, have built their careers studying the mechanisms that regulate calorie intake, expenditure, and body fatness. Schwartz has been battling the “Conventional Model” since the 1980s, as I detail in my book The Hungry Brain. Here’s an illustrative quote from my interview with him (page 145):
I started my fellowship in 1987 and was immediately indoctrinated into the idea that there is an adiposity control system. What I was working on was considered way out of the mainstream at the time. Everyone just assumed that obesity is a problem where people eat too much, and if they could control their eating and be normal, they wouldn’t have this problem.
Schwartz and others in his field proceeded to spend 30 years debunking this assumption. This field is the very reason the Conventional Model has lost steam over the last three decades. This is the field that discovered leptin and satiety hormones, showed that hormones and specific brain circuits regulate appetite and body fatness, and overturned the notion that body fatness is just about how much we happen to decide to eat and move (note that this is perfectly compatible with the prevailing view that when calorie intake is held constant, all foods are similarly fattening in humans).
Here’s another illustrative quote, this time from the abstract of the Endocrine Society scientific statement (3):
Growing evidence suggests that obesity is a disorder of the energy homeostasis system, rather than simply arising from the passive accumulation of excess weight.
It’s beyond my understanding how a person could read this paper and interpret it as a defense of the Conventional Model, or continue to believe that the Conventional Model is the primary alternative to the CIM.
In fact, our choice of models is not between the CIM and the Conventional Model, and I’m starting to feel like a broken record pointing this out. The Endocrine Society statement presents a third model, not mentioned by Ludwig and Ebbeling, that is neither the naive and oversimplified Conventional Model nor the CIM. It happens to be widely supported within the scientific community. It acknowledges many influences on body fatness, particularly the brain circuits that regulate food intake, calorie expenditure, and body fatness in response to environmental and internal signals (3). If Ludwig and Ebbeling want to convince the scientific community that their model is better than what currently exists, they should be arguing against the prevailing model, not a straw man that has been obsolete for quite some time.
Dietary fat can be fattening
This is a simple observation that is hard to reconcile with the CIM. Researchers have known for decades that adding fat to food tends to increase body fatness in animals. I have seen Olympics-worthy intellectual gymnastics to try to rationalize away this fact, but as I will show, the conclusion is impossible to escape. Here is a quote from a review paper on this topic (4): “With few exceptions, obesity is induced by high-fat diets in monkeys, dogs, pigs, hamsters, squirrels, rats, and mice.” This paper reviews many studies in these species suggesting that higher fat, usually at the expense of carbohydrate, increases body fatness.
In my own research, we used refined diets that were 60 percent fat and 20 percent carbohydrate to induce obesity in normal rats and mice– and they were very effective! In my hands, these diets caused detectable fat gain in three days, more than doubled body fat content in two weeks, and increased body fat by sixfold in 20 weeks (5, 6). The weight and fat gain on these diets is rapid, massive, and unmistakable. In fact, fat gain on refined high-fat diets occurs even when calorie intake isn’t allowed to increase, suggesting that they impact both calorie intake and expenditure (7). In their paper, Ludwig and Ebbeling suggest that only the CIM can explain the phenomenon of fat gain without increased calorie intake in rodents, but this is clearly not the case– my field has been aware of this in the context of high-fat diets and brain lesions for a long time.
These diets aren’t just high in fat of course. They’re based on refined ingredients and they usually contain carbohydrate (e.g., 20% of calories) including sugar (e.g., 7% of calories). Aha, you may say! The refined carbohydrate is why the diet is fattening, not the fat! Researchers have already tested that supposition by comparing this diet to a low-fat version in which the fat is mostly replaced by refined carbohydrate. Although the low-fat version can be fattening under some conditions (8), it isn’t as fattening as the high-fat version, and often it isn’t fattening at all. Here is a graph from a recently published study by Matthew Dalby, PhD, and colleagues, comparing fat mass gains over 8 weeks of free access to unrefined low-fat (black), refined low-fat (blue), and refined high-fat (red) food in mice (9):
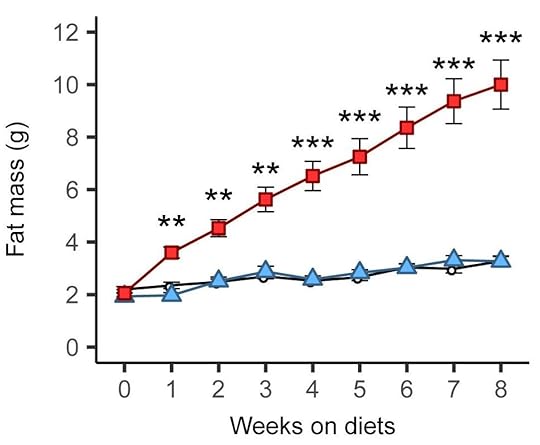
There was no difference between refined and unrefined low-fat diets, but the refined high-fat diet led to rapid and marked fat gain. This finding has been replicated independently, including in rats (10).
Refined carbohydrate and sugar are probably part of the reason why refined high-fat diets are fattening in animals, but there is no escaping the conclusion that the fat itself plays a key role. This simple observation is hard to reconcile with the CIM as articulated by Ludwig/Ebbeling/Taubes.
How about in humans? So far, two controlled overfeeding studies have compared diets rich in fat vs. carbohydrate and measured body fat gains (11, 12). Both studies reported that overfeeding with a high-fat diet produced the same, or even slightly greater, body fat gain than overfeeding with a high-carbohydrate diet of equal calories. Clearly, an excess calorie of fat gets into fat tissue as effectively as an excess calorie of carbohydrate, regardless of their effects on insulin.
How about when calorie intake isn’t controlled? Under these conditions, volunteers offered a high-fat diet tend to overeat and gain fat, just like in the animal studies discussed above (13, 14, 15, 16). Note that these studies didn’t use low-carbohydrate diets; they used high-fat diets that were unrestricted in carbohydrate, which is presumably why the results differ from low-carbohydrate diet studies in which people tend to eat fewer calories and lose fat.
Historically, many cultures ate high-carbohydrate (and high-glycemic-load) diets and were lean
This is another simple observation that is difficult to reconcile with the CIM. More precisely, it is a collection of many observations, because there have been, and continue to be, countless lean high-carbohydrate cultures. Ludwig has responded to this in the past, arguing that 1) the data are of insufficient quality to draw conclusions, 2) these people were lean because they were on the verge of starvation, and 3) they were protected by high levels of physical activity (17).
I’ll address these in turn, starting with the assertion that the data are of insufficient quality. The quality of data vary across observations, but some are quite high. For example, in 1990, Staffan Lindeberg, MD, PhD conducted a detailed survey of the diet, lifestyle, and health of the residents of Kitava, a Melanesian island scarcely touched by industrialization. He found a diet based on starchy plant foods (African yam, sweet potato, taro, cassava), fruit, vegetables, seafood, and coconut, with 69 percent of calories coming from carbohydrate (compared to ~49% in the US currently) (18).
Lindeberg described the Kitavans as “characterized by extreme leanness (despite food abundance)” (18). And the data back it up. Of the 247 Kitavans Lindeberg examined, none had obesity, and only a few were on the cusp of overweight (18). They did not gain weight with advancing age. There was no diabetes. Lindeberg described to me how the Kitavans had so much food they would let much of it rot or feed it to their animals. This is not to say that they never experienced food shortages– like all traditionally-living cultures, they probably did sometimes. However, they were not experiencing food shortage at the time of Lindeberg’s visit, and this is obvious in the photos he took of them (photos courtesy of Staffan Lindeberg):



Lindeberg actually did observe two Kitavan men who had borderline obesity. He describes them in his book Food and Western Disease (p. 119):
At the time of our survey in Kitava, we observed two cases of abdominal obesity, both in urbanised male migrants who had grown up in Kitavan and now came for a visit. We managed to examine one of them, a businessman aged 44 who differed in four variables from all other adults regardless of sex: he had the highest [body mass index], the highest waist-to-hip ratio, the highest diastolic blood pressure, and the highest PAI-1 in blood plasma… The most obvious difference in his lifestyle, as compared with non-migrant Kitavans, was the adoption of Western dietary habits.
This suggests that Kitavans were not genetically protected from fat gain and its metabolic consequences– they were protected by their diet/lifestyle, despite its high glycemic load. When they adopt an industrialized diet, which happens to be higher in fat and lower in carbohydrate, they become fat just like the rest of us.
Were the Kitavans protected from obesity by very high levels of physical activity? Lindeberg tested this hypothesis, and here’s what he found (18):
The amount of physical activity is estimated at 1.7 multiples of the basal metabolic rate, which is slightly higher than in sedentary Western populations.
In Food and Western Disease, he states that this corresponds to Westerners with “a moderate amount of physical activity on the job and during leisure time” (p. 84). I have little doubt that regular physical activity is part of the reason why Kitavans were lean and healthy, but their physical activity level was nothing extreme.
The book Western Diseases: Their Emergence and Prevention is primarily a collection of field studies from around the world that document the transition between traditional and modern lifestyles, and its health impacts. In most (not all) cases, the transition was from a very-low-fat, very-high-starch, high-glycemic-load diet to a more fatty diet, and this was invariably accompanied by increased rates of obesity and chronic disease. Certainly, the shift from carbohydrate to fat wasn’t the only change and we can’t attribute increasing body fatness to it exclusively. Concurrently, many of these cultures experienced increased consumption of sugar, salt, alcohol, and lower physical activity, among many other changes. These observations are nevertheless hard to reconcile with the idea that the CIM is the primary explanation for body fatness.
Just to ensure that there is no room to dispute the point I’m making here, I’ll discuss one more culture: Japan. Western Diseases contains historical data on the Japanese diet between 1950 and 1975, a time during which obesity was uncommon in Japan (p. 338). In 1975, long after postwar food shortages were over, the Japanese diet was 62 percent carbohydrate, with most of that carbohydrate coming from white rice. Furthermore, the predominant type of rice was high-amylopectin sticky rice, as it still is today (remember this word amylopectin, we’ll come back to it later). This type of rice has one of the highest glycemic loads of any food– precisely what Ludwig and Ebbeling argue is the most fattening. The Japanese ate it daily as their primary staple food, yet the prevalence of overweight and obesity were, and continue to be, lower than any other industrialized nation (19). As in many other cultures, over time the Japanese diet has become less reliant on rice and more reliant on fats, meats, sweets, and other foods, and obesity rates have increased, although they remain low compared to other countries.
Are Japanese people genetically resistant to obesity? Clearly not, because when they emigrate to the US they become much heavier (20, 21). Japanese people living in Japan are protected by their diet and lifestyle habits, despite the high glycemic load of their diet. What is different about the Japanese vs. Japanese-American diet? Many things, but here’s an obvious one: “Although total caloric intake was not greatly different between Japan and Hawaii, the percent caloric intake as fat was two times greater in Hawaii” (22). When they emigrate and their diets and lifestyles Americanize, they tend to develop overweight and obesity like people of European, African, and Native American descent, despite a reduction in dietary glycemic load.
To be clear, I’m not arguing that increased fat intake is the primary reason Japanese-Americans are heavier than Japanese, and Japanese today are heavier than in 1975– the point is to demonstrate that declining carbohydrate intake and glycemic load corresponded with an increase in body fatness. The CIM cannot be the primary explanation for obesity trends in this case, and I would argue, many other similar cases.
The genetics of common obesity argues against insulin and fat cells as the central mechanism of fat gain
Obesity has a strong genetic component, and geneticists are making great strides in cracking its code. As it turns out, the genetics of body fatness is incredibly complex, with differences at probably thousands of locations in the genome contributing to it. Each location only has a very small effect, but together they add up to a large effect. The latest and largest genome-wide analysis in 700,000 people has identified 716 places in the genome where genetic differences impact body fatness (23). By examining the functions of the implicated genes, we can gain insight into the mechanisms that determine body fatness. We know this method works: as expected, genes impacting type 2 diabetes risk tend to relate to insulin secretion, insulin sensitivity, and the pancreas, and genes impacting height tend to relate to skeletal and connective tissue growth (23, 24).
For our purposes, a key aspect of these studies is that they are unbiased (not hypothesis-driven). Researchers simply scan the entire genome looking for associations and report whatever they find, regardless of whether it conforms to our pre-existing beliefs about obesity. Whatever the mechanisms are that contribute to common obesity, they should turn up.
So what mechanisms do these studies report as being most intimately linked to obesity? Are they genes related to fat cell function? Insulin signaling? In fact, these studies consistently report that obesity-linked genes relate primarily to brain development and function: “[body mass index]-associated genes are mostly enriched among genes involved in neurogenesis and more generally involved in the development of the central nervous system” (25). This is consistent with the prevailing view in my field that the brain is in the driver’s seat, not insulin or the fat cell. That doesn’t mean the brain is the only thing that matters– probably a lot of things matter to some degree– but the brain appears to be the most important driver of common obesity. This is why my book is titled The Hungry Brain and not The Hungry Fat Cell.
I can’t overstate the importance of these genetic findings for evaluating hypotheses about obesity. They offer a rare and valuable, unbiased, 10,000-foot view of the mechanisms that drive excess body fat accumulation in the general population. We should be calibrating to these findings and asking ourselves hard questions if our hypotheses are inconsistent with them.
Circulating free fatty acids are not reduced during active fat gain in rodent models of dietary obesity
Another serious problem for the CIM is that people with obesity have circulating levels of glucose and free fatty acids (the two main circulating fuels) that are normal or elevated– not reduced as the CIM predicts (26, 27, 28, 29). Despite high levels of circulating insulin in most people with obesity, neither insulin nor anything else is preventing the release of fat from fat cells. To address this problem, Ebbeling and Ludwig conceive of a novel concept they call the “dynamic stage of obesity development”. According to this concept, during the “dynamic stage”, people are actively gaining weight because insulin is shunting fat into their fat cells, suppressing circulating fatty acids levels. But after the dynamic stage is over, weight plateaus and circulating glucose and fatty acid levels normalize or increase, which is presumably why, despite countless studies, the dynamic stage has never been observed.
Thus the CIM hinges on a phenomenon proposed by Ludwig and Ebbeling that has never been observed, and that honestly I find a bit too convenient. But has it been disproven? Let’s see if we can find any data. What we want are time course studies where researchers measure circulating free fatty acid levels at regular intervals in humans or animals that are actively gaining fat. I found two studies in rats, both of which report that circulating free fatty acids were normal or elevated at all points during active fat gain resulting from a fattening diet (30, 31). In this model of obesity at least, there is no “dynamic stage of obesity development” that supports the predictions of the CIM. Importantly, obesity was caused by a fattening diet in these studies. This is more pertinent to common human obesity than studies that involve brain lesions, leptin deficiency, or excess insulin injections, which tend to be cited by Ludwig and Ebbeling.
Reducing the flow of fatty acids out of fat cells doesn’t reduce energy expenditure, increase food intake, or increase body weight in humans
The cornerstone of the CIM is the idea that insulin suppresses fat release from fat cells, reducing energy levels in the blood, which reduces metabolic rate (energy expenditure), increases hunger, and causes fat gain. What if there were a way to experimentally restrict the flow of fatty acids out of fat cells, independently of insulin? If the CIM is correct, we should see a decrease in energy expenditure, an increase in food intake, and fat gain.
Fortunately for us, there is a way to do this: a drug called acipimox. Acipimox inhibits lipolysis, or the release of fatty acids from fat cells, mimicking the effect of insulin (32). As a consequence, free fatty acid levels in circulation decline. Acipimox has been used in humans many times, but of particular interest to us is a six-month randomized controlled trial that reported the impact of acipimox vs. placebo on energy expenditure, food intake, and body composition (33). The (preregistered) primary goal of this study was to examine the effects of acipimox on mitochondrial function, but it reported a number of other outcomes (34).
Researchers randomly assigned 39 men and women with obesity to acipimox or placebo for six months. 31 continued for the whole 6-month period (16 in acipimox and 15 in placebo) and compliance was excellent. Acipimox robustly and consistently inhibited lipolysis and at the six-month time point fasting free fatty acids in circulation were reduced by 38 percent.
How did energy expenditure change? “No significant effects of acipimox compared to placebo on [resting energy expenditure] or [respiratory quotient] were observed during either the fasting state or the hyperinsulinemic clamp.” And: “Changes in physical activity over 6 months were not different between groups”
How about food intake? According to 4-day food records: “Caloric and relative macronutrient intake did not change significantly between groups”.
Body composition? To measure this, they used the accurate DEXA technique: “There were no significant effects of acipimox vs placebo on measures of body composition, including [body mass index], [visceral adipose tissue], or lean body mass”.
Acipimox simulated the effect of insulin on fat cells that is supposed to make us hungry and fat according to the CIM, but it had no effect on food intake or body composition. Furthermore, this study suggests that the brain doesn’t respond to low free fatty acid levels by increasing food intake, which is another unproven assumption that underlies the CIM. These findings are very hard to reconcile with the CIM.
Animal studies cited by Ebbeling and Ludwig are not as supportive as suggested
Ludwig and Ebbeling cite rodent studies their team and others conducted in which they placed rats and mice on low-glycemic/insulinemic vs. high-glycemic/insulinemic diets. Since they rely heavily on these rodent studies in their arguments, and ostensibly believe rodents are useful animal models for human obesity, let’s take a closer look at them.
Let’s start with a series of studies, conducted by Gerard Slama and colleagues between 1996 and 1998, cited by Ludwig and Ebbeling. These compared diets based on mung bean noodles (low glycemic) or ground-up toast (high glycemic) in rats. You read that correctly: mung bean noodles vs. powdered toast. According to the authors, mung bean noodle starch is 8 percent indigestible fiber (resistant starch) while ground-up toast starch is only 2 percent indigestible fiber (35). So right off the bat, these diets were not isolating the glycemic index as a variable– they also differed substantially in fiber content, somewhat in calorie density, and probably in many other ways. Furthermore, resistant starch is a type of fiber that is actively under study for its potential weight and health benefits.
In the first study, published in 1996, the researchers put diabetic and nondiabetic rats on the two diets for five weeks (36). I’ll let them share the results in their own words: “Body weight was significantly lower in rats (normal and diabetic) fed on the mung-bean starch diet until the fourth week. However, body weight was comparable for both diets at the end of the 5-week period.” I don’t see how this supports the CIM.
Onward to the second study, published in 1998 (37). Again, they put diabetic and nondiabetic rats on the noodles vs. toast diets, this time for three weeks. The abstract says what we want to know: “After 3 wk, food intake, epididymal fat pad weights, and plasma glucose, insulin and triglyceride concentrations did not differ between diet groups.” Again, I don’t see how this supports the CIM.
The third study was published later in 1998 (38). Same design and the previous study, same findings: “After 3 wk, neither body weights nor relative epididymal fat pad weights differed.” None of the three studies cited by Ludwig and Ebbeling found that noodles vs. toast meaningfully impacts weight or body fatness in rats, despite other differences in diet composition that should have favored the low-glycemic noodle diet.
Let’s move on to a study conducted by Ludwig’s group and published in 2004 (39). It’s much better controlled than the noodles-vs.-toast studies, as the only difference between the two diets was the type of starch: fast-digesting amylopectin vs. slow-digesting amylose. In the first experiment, the researchers began by removing 60 percent of the rats’ pancreas to impair their insulin secretion. I don’t buy the rationale for doing this (wouldn’t reducing insulin secretion make carbohydrate less fattening according to the CIM?); they say it’s to make the rats more similar to people with prediabetes. Rats were fed the diets for 18 weeks. The amylopectin group began gaining weight halfway through and the researchers had to restrict their food intake to keep their weight similar to the amylose group. Despite no differences in weight and a lower calorie intake, the amylopectin group ended up fatter than the amylose group at the end, although neither group was especially fat (18 vs. 10 percent body fat).
I’m going to skip over the second experiment because it isn’t very relevant here. In the third experiment, they fed normal obesity-prone mice (C57BL/6J strain) the two diets for 9 weeks without restricting their intake. Body weight didn’t differ at the end, but body fatness was meaningfully higher in the amylopectin group (25 vs. 14 percent body fat). A longer-term study by Ludwig’s group also showed that an amylopectin-based diet leads to a meaningfully higher level of body fatness than an amylose-based diet in mice (40).
Together, these experiments do suggest that amylopectin is more fattening than amylose in rodents, although amylopectin-fed rodents look like runway models next to those fed refined high-fat diets (as described above). However, it’s strange to me that they only reported results from rats that had 60 percent of their pancreas removed. No matter, because I was able to find another study that put normal rats on amylose or amylopectin diets for 16 weeks (41).
This study is really interesting because it not only included amylose and amylopectin-based diets, it also included a third glucose-based diet. Glucose is digested and absorbed even more quickly than amylopectin, and stimulates insulin to a correspondingly greater degree. So if the glycemic index is what really matters, we should see the most weight gain in the glucose group, followed by the amylopectin group, followed by the amylose group. Here’s what happened: “There was no significant difference in body weight between the [amylose] or [amylopectin] groups at any of the testing points. The [glucose]-fed animals gained weight at a slightly lower rate than the [amylose] or [amylopectin]-fed animals. The [glucose]-fed animals had a significantly lower body weight than [amylose]- or [amylopectin]-fed animals at both the 8- and 16-wk testing points.” This is despite elevated insulin secretion in the glucose group.
The highest-glycemic diet caused the least weight gain, and there was no difference between the amylopectin and amylose groups eating ad libitum, in contrast to Ludwig’s study where they had to restrain the amylopectin-fed rats’ food intake to rein in body weight. It’s worth repeating that 16 weeks of a refined high-fat diet causes severe, unmistakable obesity in rodents (42, 43).
This leads me to question whether glycemic/insulinemic index is the relevant difference between amylose and amylopectin-based diets. If that were the case, wouldn’t a glucose-based diet be the most fattening of all? Another thing that leads me to question the relevance of these findings is the human evidence. Low-glycemic/insulinemic diets are not very effective for fat loss in humans, including in Ludwig and Ebbeling’s randomized controlled trial where their low-glycemic diet caused weight loss nearly identical to a low-fat diet (44). Then there are the Japanese data I discussed above, where the consumption of amylopectin-rich sticky white rice as the primary staple food did not lead to obesity (Western Diseases: Their Emergence and Prevention).
Is the CIM well-enough supported to justify diet advice?
Ludwig and Ebbeling state that “high-quality research will be needed to resolve the debate”, yet their paper contains a panel titled “Dietary Recommendations Based on the Carbohydrate-Insulin Model”. As I recall, one of the favorite pastimes of CIM advocates is criticizing the USDA for prematurely dispensing low-fat diet advice in the 1980s. If the science isn’t settled yet, perhaps it’s not the right time for bestselling books that confidently promote the CIM and a diet based on it to the public?
Conclusions
The question we must answer is not “can we find evidence that supports the CIM”, but rather “does the CIM provide the best fit for the totality of the evidence”. Although it is certainly possible to collect observations that seem to support the CIM, the CIM does not provide a good fit for the totality of the evidence. It is hard to reconcile with basic observations, has failed several key hypothesis tests, and currently does not integrate existing knowledge of the neuroendocrine regulation of body fatness.
Certain forms of carbohydrate probably do contribute to obesity, among other factors, but I don’t think the CIM provides a compelling explanation for common obesity.
Tweet
Share on Tumblr
June 20, 2018
Nutrition Science Initiative (NuSI) in retrospect
On Monday, Wired published an article about the Nutrition Science Initiative, “The collapse of a $40 million nutrition science crusade“. This seems like a good time to review the trajectory of NuSI and what we can learn from it. NuSI was founded in 2012, ostensibly on the idea that our current understanding of obesity is at best incomplete, and possibly incorrect, and we need larger and more rigorous experiments to tease out its true causes. In practice, it was founded to investigate the carbohydrate-insulin hypothesis of obesity, the favored hypothesis of its co-founder Gary Taubes (its other co-founder is Peter Attia, MD). It was clear in early materials released by NuSI that its staff believed strongly in this hypothesis and felt that these experiments would vindicate it and address the obesity crisis. They predicted their efforts would reduce the prevalence of obesity by 50 percent, and the prevalence of diabetes by 75 percent, by 2025.
These grand ambitions, coupled with Taubes’s high-profile public advocacy stretching back to 2002, and Attia’s energy and vision, netted NuSI over 40 million dollars, almost all of it from the Arnold Foundation. In 2012, I was contacted by Taubes and Attia, who explained what NuSI was and asked for my support. As I explained in a post I wrote at the time, I ended up endorsing NuSI because I viewed it as an additional funding mechanism for testing questions related to obesity and nutrition that may otherwise be difficult to fund. Equally important, I saw that they were selecting high-quality researchers to do the experiments rather than cherry-picking people who were on the home team. I had a conversation with Kevin Hall, PhD, during which he reassured me that under the terms of the agreement, NuSI wouldn’t have the ability to tinker with study data or interpretation. More funding for interesting science, and an agreement that prevents scientific tampering. What could go wrong?
That said, I did express major reservations about NuSI at the time. Their materials suggested that they were heavily biased, and, to quote myself, “It would be remiss of me not to question the wisdom of putting a major science funding mechanism into the hands of a journalist who is, shall we say, very attached to his ideas.” Despite these reservations and my substantial uncertainty about whether NuSI would support good science or become a corrupting influence, I felt that it had more potential to do good than harm.
The first study NuSI funded was a meticulous eight-week pilot trial, led by Hall, to see if a very-low-carbohydrate (ketogenic) diet causes a meaningful increase in calorie expenditure vs. a calorie-matched standard American diet with more than tenfold the sugar. Volunteers were kept in a research facility where every morsel they ate was controlled. The study design was agreed upon in advance by both the researchers and NuSI, and the diets were designed by low-carbohydrate diet researcher Jeff Volek, MD. According to Taubes, calorie intake is virtually irrelevant and insulin is everything, so his hypothesis predicts large and obvious differences in calorie expenditure and fat loss between groups, despite equal calories. The results showed little effect of the ketogenic diet on calorie expenditure, and an initial slowing of fat loss, despite a large and persistent decline in insulin secretion.
NuSI reacted poorly to this news. They suddenly became very critical of the study’s design, and according to a timeline shared by Hall, “requested access to the data and began providing extensive comments on the data analyses and interpretation”. NuSI also requested that the researchers withdraw an abstract on the study they had submitted for the American Diabetes Association meeting, which they did. I find this particularly galling.
The study did have substantial limitations, most notably its lack of a true control group*. After seeing the study’s results, Taubes began arguing that these limitations make the study uninformative, reversing his initial support for its design. The truth is that, despite the study’s limitations, there is no way to reconcile its findings with Taubes’s version of the carbohydrate-insulin hypothesis. If carbohydrate and insulin do what Taubes thinks they do, the extreme difference in carbohydrate and sugar intake between the two experimental diets would have made this clearly visible. NuSI seemed to understand this before the results came in.
The interference from NuSI became so intolerable that in April of 2015, Hall and the other researchers wrote an email to NuSI asking to “reestablish the academic freedom” of the research team, as per their initial agreement. Rather than backing off, NuSI doubled down. In June of 2015 it “confirmed that it wanted to be more closely involved in the design, analysis, and interpretation” of the next study. This is documented in Hall’s timeline.
From there, NuSI entered free fall. Attia resigned in December of 2015. After a spat between Hall, Taubes, and Mark Friedman, MD in front of John Arnold in January of 2016, which presumably broke the camel’s back, Hall resigned from his NuSI-supported position. The Arnold Foundation stopped sending checks, and NuSI withered. NuSI still exists today, but most of its staff are gone and it’s limping along seeking funding.
In retrospect, it’s not surprising that NuSI collapsed under the weight of its own ideology. The nature of ideology is that it’s inflexible when it encounters facts that contradict it**. It cannot bend, it can only break.
One thing that I think hasn’t gotten enough attention is the fact that NuSI staff, particularly Attia, paid themselves enormous salaries while they were at NuSI***. According to public records, Attia received an average of $425,000 per year for four years, including $727,754 in 2015, for a grand total of $1.7 million. Taubes received an average of $117,000 per year for five years, netting $586 thousand. Friedman received over $200,000 per year in 2014 and 2015, netting $495 thousand.
NuSI is a 501(c)(3) nonprofit corporation, meaning that its funding is explicitly intended to benefit society, not individuals. People have to make a living of course, but it’s hard to understand the rationale for a $728k salary at a nonprofit. Although Taubes’s salary appears more reasonable than Attia’s, he was writing and promoting The Case Against Sugar and doing other non-NuSI work during this time, so it appears to be for part-time work. I’m not going to come to any grand conclusions about this before hearing their side of the story, but they certainly have some explaining to do.
Here are my takeaways from the NuSI experiment, with the benefit of hindsight:
Obesity is a difficult problem to solve. Many grand ambitions have foundered on this fact.
As a result of NuSI’s funding, useful research was published, and more potentially useful research is forthcoming. So I don’t think it’s correct to view NuSI as a failure, despite the fact that its funding and staff have mostly evaporated.
Certain important norms in science only work as long as everyone is behaving themselves, and this makes science fragile.
Ideology and science don’t mix. The risk in giving ideologues power is not simply that they may fail, but that they may fundamentally corrupt science.
* One could argue that the subjects were their own controls, but ideally for this to work it should be done in counterbalanced crossover fashion with a wash-out period between diets.
** Philip Tetlock, PhD describes ideologues beautifully in his book Superforecasting. He refers to them as “hedgehogs” and pragmatists as “foxes”. Hedgehogs are persuasive because in their eyes, the world is simple and they are utterly convinced they know how it works. But they are abysmally bad at making predictions because the world isn’t as simple as they think. Most television pundits are hedgehogs and their predictions are about as good as flipping a coin. Foxes, on the other hand, understand that the world is complex and try to factor this complexity into their worldview. They learn from their mistakes. As a result, they are better at making predictions. But they get less attention because they’re less confident and their view of the world is harder to explain.
*** I initially learned about this in a post by Evelyn Kocur. I don’t endorse the aggressive tone of the post but she certainly did her homework. I have independently confirmed the salary figures in her post.
Tweet
Share on Tumblr
May 24, 2018
What is a low-glycemic diet, really?
 I’ve always been struck by the imprecision of how the terms “low-glycemic-index diet” and “low-glycemic-load diet” are used. In theory, these are diets that reduce post-meal blood glucose and their benefits derive from this property. In practice, they often differ from typical diets in multiple ways. If a “low-glycemic diet” is also higher in fiber and protein and/or lower in palatability, calorie density, and carbohydrate, can we really attribute its beneficial effects to its impact on blood glucose per se? To gain a better understanding of the depth and pervasiveness of this problem, I did a scientific literature search for randomized controlled trials of low-glycemic-index/load diets, selected the first ten I found, examined the details of the assigned diets, and evaluated the findings.
I’ve always been struck by the imprecision of how the terms “low-glycemic-index diet” and “low-glycemic-load diet” are used. In theory, these are diets that reduce post-meal blood glucose and their benefits derive from this property. In practice, they often differ from typical diets in multiple ways. If a “low-glycemic diet” is also higher in fiber and protein and/or lower in palatability, calorie density, and carbohydrate, can we really attribute its beneficial effects to its impact on blood glucose per se? To gain a better understanding of the depth and pervasiveness of this problem, I did a scientific literature search for randomized controlled trials of low-glycemic-index/load diets, selected the first ten I found, examined the details of the assigned diets, and evaluated the findings.
Methods
I performed a search in Google Scholar using the search terms “randomized trial low glycemic”. I scanned down the list until I had identified the first ten studies that met the following criteria:
Randomized controlled trial
Compares a low-glycemic-index or low-glycemic-load diet to a control diet
Title describes the low-glycemic diet only according to its glycemic index/load, not other diet characteristics (e.g., high protein, legumes)
Any length (single meal to long-term diet trial)
I extracted the following information from each study:
First author
Year published
Title
Summary of subject characteristics
Length of intervention
Diet summary
Detailed diet description
Was the between-diet comparison well controlled for glycemic index/load? Yes = differences in glycemic index/load were achieved without substantial differences in the types of foods consumed. Moderate = the types of foods differed substantially between groups, but major nutritional characteristics like protein and fiber intake were well controlled. No = major nutritional characteristics like protein and fiber intake differed between groups.
Outcome of the intervention
My primary outcome was how well each diet was controlled for dietary variables other than glycemic index/load.
Results
I compiled a table of the findings here. Trials were published between 2004 and 2011, and were all highly cited (105 to 413 citations according to Google Scholar). They lasted between 4 weeks and 18 months, and 6 of 10 involved subjects with diabetes.
Among the ten trials, five were clearly not controlled for glycemic index/load (“no”), four were moderately well controlled (“moderately”), none were well controlled (“yes”), and one didn’t provide enough information to judge.
Overall, the results of the trials suggest that low-glycemic diets can be helpful for blood glucose control in diabetes, but their effects are modest in people without diabetes. They are not very effective for weight or fat loss (relative to control diets, which varied) and they seem to have little impact on birth outcomes in the context of gestational diabetes.
Discussion
In this limited but unbiased sample of highly cited papers on low-glycemic diets, I found that none isolated glycemic index/load as a variable, 40 percent were moderately well controlled for glycemic index/load, and 50 percent were poorly controlled for glycemic index/load. Another way of stating this is that all of the diet comparisons differed in ways other than glycemic index/load, and five of them differed in major ways that are likely to be confounding.
This is only a problem if we attribute the outcomes of these studies to glycemic index/load per se. If we all understand that “low-glycemic diet” often means “beans, nuts, fresh fruit, and slow-digesting whole grains that may improve health via multiple mechanisms relative to typical diets”, there’s no problem. This is the interpretation I believe is most accurate, but it’s not what I see in most discussions of these studies.
The findings of this brief literature survey suggest that we should be cautious about attributing the effects of low-glycemic diets to their impact on glycemia per se.
Tweet
Share on Tumblr
April 1, 2018
My new favorite juicer
Let’s face it: most juicers are terrible. They’re complicated, hard to clean, expensive, and they break down. Worst of all, from a nutritional standpoint they waste important nutrients, especially fiber. What if there were a juicer that solved all these problems at once? I’ve finally found it: the IllaMAX 3000.
The IllaMAX 3000 juices apples, celery, berries, kale, and all my favorite nutrient-packed foods with ease. It can even make smoothies! You get the delicious juice, plus all the nutritional benefits of the fiber. And you don’t end up having to throw away the leftover pulp because all it’s in the juice. How can this be possible?
Two other remarkable facts about the IllaMAX 3000. First, unlike other juicers, it’s self-cleaning, so you’ll never have to scrub your juicer again. Second, it’s so incredibly durable it can last up to 122 years.
This might seem too good to be true, but it’s made possible by some very advanced technology:
Rock-hard hydroxylapatite grinding stones are built for a lifetime of use
Two powerful mechanical levers apply crushing and grinding force to the stones
A reliable peristaltic pump collects the juice
 A diagram of some of the key components of the IllaMax 3000
A diagram of some of the key components of the IllaMax 3000You won’t believe the price. Not $199.99. Not $99.99. Not even $49.99. But absolutely FREE! In fact, most people don’t realize that they may even already have one installed!
 “The IllaMAX 3000 is a beast!” -Mark Twain
“The IllaMAX 3000 is a beast!” -Mark TwainApril Fools!
Tweet
Share on Tumblr
March 25, 2018
Microbiota and obesity: Is it all hype?
On the Amazon page for my book The Hungry Brain, the top review is positive overall but regrets that I didn’t cover the role of the gut microbiota in obesity. This reflects a common belief that differences in the microbial composition of the human gut are an important determinant of body fatness. In fact, the omission in my book was deliberate, because despite the popularity of this idea, I’m still not convinced it’s correct. In this post, I’ll review three recent findings that have added to my skepticism, including a human fecal microbiota transplant study that doesn’t mean what most people think it means. I’ll note up front that I don’t think this evidence proves that the gut microbiota isn’t involved in determining body fatness– it just proves that the hype is outpacing the science.
Finding #1
The first finding is actually a reanalysis of a seminal result in the microbiota-obesity field. The initial paper was published in the journal Nature in 2006, and to large extent, it built the field. The key experiment shows that a fecal microbiota transplant from an obese mouse to a lean (microbiota-free) mouse can make the latter gain fat. This was an important milestone because it demonstrated for the first time, apparently unequivocally, that the microbiota was actually causing part of the fattening effect– not just along for the ride. The study has been cited 5,931 times.
I’ve always been skeptical of this paper, for different reasons, but recently a microbiome researcher named Matthew Dalby took a closer look at the data underlying the claim that body fatness can be transferred via the microbiota and published the results on his blog. I won’t get into the details of his analysis– you can read them on his blog if you’re interested– but suffice it to say the finding doesn’t survive critical inquiry. The effect size is trivial and it’s likely explained by a statistical artifact called regression to the mean. This type of anomaly is one of the hazards of using small groups of animals in your experiments (9 – 10 per group in this case), and it was exacerbated by the way the researchers chose to report the data.
This undermines a seminal finding of the microbiota-obesity field and increases my skepticism about the hypothesis as a whole.
Finding #2
The second finding is also from Matthew Dalby, but this time in the form of a study. Published late last year in Cell Reports, the study compares body fatness, metabolic health, and gut microbiome of mice fed three different diets:
Unrefined low-fat diet. This is the typical maintenance diet for rodent colonies and it’s primarily composed of unrefined grains and soybeans.
Refined low-fat diet. This diet is composed mostly of refined carbohydrate, largely corn starch and maltodextrin, but also a bit of sucrose. It contains casein for protein and a few other ingredients.
Refined high-fat diet. This is the typical style of diet that’s used to produce obesity in rodents, including in my own experiments. It’s mostly lard by calories, but also contains corn starch, maltodextrin, and a bit of sucrose. It contains casein for protein and a few other ingredients.
There are many interesting findings in this paper, but for our purposes there is one key result. The mice fed the unrefined low-fat diet and the refined low-fat diet remained lean and metabolically healthy (good glucose tolerance), while the mice fed the refined high-fat diet quickly became obese and metabolically unhealthy (poor glucose tolerance)…
…Yet when they examined the gut microbiota, it was similar in the mice eating the refined low-fat and refined high-fat diets (while the microbiota on both refined diets were quite different from the unrefined diet). Therefore, differences in the microbial composition of the gut flora couldn’t explain differences in body fatness or metabolic health.
Finding #3, or how not to analyze data and report study results
The ultimate proof that gut microbiota impacts human body fatness would be a study demonstrating that a fecal microbiota transplant from lean people to people with obesity can cause weight loss, or vice versa. The first study to examine the weight impact of a fecal microbiota transplant from lean people to people with obesity was recently published— let’s have a look.
The first place we’ll go is to the study’s preregistration site. This is the place where the authors laid out their research plan prior to conducting the study, including how the data would be analyzed an interpreted. The reasons authors preregister research plans, aside from the fact that it’s often required by quality journals, is that it shows that they didn’t tweak their analysis and reporting after seeing the data, in order to engineer a preferred outcome. This can lead to results that are convenient for the researchers but misleading for the scientific community. Preregistration is a critically important tool for combating the “replication crisis” that is currently sweeping many fields of research as we realize their findings aren’t as reliable as we thought they were.
One of the most important things a researcher should preregister in any randomized trial is the study’s “primary outcome”. This is the outcome that will matter the most for study design, data analysis, and interpretation. It is the outcome that researchers commit in advance to focusing on in the resulting publication, no matter what the results are. Researchers can also register “secondary outcomes”, which are less important. In this case, the preregistered primary outcome was “changes in weight in relation to fecal flora composition and short chain fatty acid metabolism in fecal samples after 3, 6, 12 and 18 weeks”. This isn’t very specific but it’s clear that the primary outcome is about changes in body weight. Thus, according to the researchers themselves, the study was designed and committed in advance to be primarily about body weight. Insulin resistance is listed as a secondary outcome.
Onward to the paper. The title should immediately send up a red flag: “Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition”. The title focuses on a secondary outcome but doesn’t mention the primary outcome of body weight, and the abstract does the same. In fact, the paper barely mentions that the intervention had no impact on body weight at the two reported time points of 6 and 18 weeks, and all the figures relating to it are in the supplementary materials. Let’s not be misled by this sleight of hand: the study was primarily about the impact of fecal microbiota transplant on body weight, and it found no effect.
The paper does claim that lean microbiota improved insulin sensitivity at 6 weeks, but this result is unconvincing due to poor statistical methods. I’ll relegate my discussion of this to a footnote so I don’t bore you with technical details.* I don’t know what the reviewers were smoking while reading this paper but it must have been strong stuff.
Conclusion
We still don’t have compelling evidence that differences in the composition of the gut microbiota significantly impact body fatness in humans, and the best human evidence we have suggests that it may not be important. New evidence also suggests that it may not be as important in rodents as we thought either. That said, the case isn’t closed yet. The human evidence we have is short-term, and given the staggering complexity of the microbiota and how it interacts with diet and lifestyle, there is still room for it to be important. I look forward to further research on it, but in the meantime, let’s cut back on the hype.
Thanks to Matthew Dalby for his thoughts on this post.
* They used within-group comparisons to show that the group receiving the lean-type microbiota showed increased insulin-mediated glucose uptake over the 6-week period, but the control group didn’t. They interpreted this as showing that the fecal transplant improved insulin sensitivity. The problem with this is that key statistical tests in randomized controlled trials have to directly compare between groups, not within groups. Otherwise, why bother having a control group?
Tweet
Share on Tumblr
Stephan Guyenet's Blog

- Stephan Guyenet's profile
- 40 followers

